Reproducible, scalable, and shareable analysis pipelines with bioinformatics workflow managers | Nature Methods
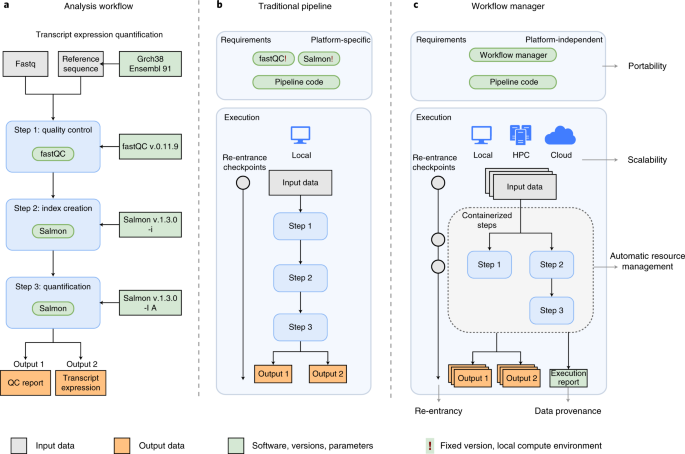 | The rapid growth of high-throughput technologies has transformed biomedical research. With the increasing amount and complexity of data, scalability and reproducibility have become essential not ... www.nature.com |
https://github.com/GoekeLab/bioinformatics-workflows
Workflow managers provide an easy and intuitive way to simplify pipeline development. Here we provide basic proof-of-concept implementations for selected workflow managers. The analysis workflow is based on a small portion of an RNA-seq pipeline, using fastqc for quality controls and salmon for ... github.com |