
Wednesday, 13 November 2013
Consumer Grade HDD are OK for Data backup
Ah storage, who doesn't need more of it? Cheaply I might add.
The folks at Backblaze published their own field report on HDD failure rate which is interesting for any data center.
Earlier I had read about Google's study on how temperature doesn't affect HDD failure rate and promptly removed the noisy HDD cooling fans in my Linux box.
Their latest blog post at http://blog.backblaze.com/2013/11/12/how-long-do-disk-drives-last/ has me thinking that some of my colleagues elsewhere that are doing Backblaze like setups should switch to consumer grade HDDs to save on cost.
I do have a 80 Gb Seagate HDD that has survived the years. Admittedly I am not sure what to do with it anymore as it is too small(80 Gb) to be useful and too big (3.5") to be portable. It was used as a main HDD until it's size rendered it obselete hence it's sitting in a USB HDD dock that I use occasionally.
maybe you can find out the age by looking up the serial number but I use the SMART data info that you can see from the Disk Utility in Ubuntu.
However the age as you can see from the screen shot is an estimate of the days it has been powered on.
Hmm pretty low mileage for a 80 Gb HDD eh?
Check out this 320 Gb IDE HDD
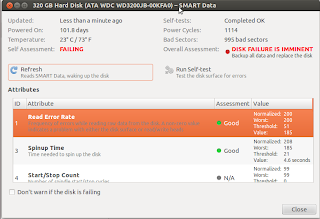
Even lower mileage! Not too sure of the history of this drive so can't really comment here.
Completely anecdotal but I have 3 Seagate 1 Tb HDD dying within a year from a software 5x HDD RAID array from within CentOS. When I checked on the powered on days it says it has been running for 3 years. So I am
1) confused how SMART data records HDD age
2) in agreement with Backblaze that HDD have specific failure phases. (where usage patterns play less of a role perhaps)
3)guessing that most of the data on Backblaze are archival in nature i.e. write once and forget until disaster strikes. So it would be great if Backblaze can 'normalize' the lifespan of the HDD with data access patterns per HDD to make it more relevant for a crowd that has a slightly different usage pattern than pure data archival needs.
That said I think it's an excellent piece of reading if you are concerned about using consumer grade HDD. Kudos the Backblaze team who managed to 'shuck' 5.5 Petabytes of raw HDD to weather the Thailand crisis (wonder how that affected their economics of using consumer grade HDD)
As usual YMMV applies here. Feel free to use consumer grade HDD for your archival needs but be sure to build in redundancy and resilience into your system like the folks in Backblaze.
The folks at Backblaze published their own field report on HDD failure rate which is interesting for any data center.
Earlier I had read about Google's study on how temperature doesn't affect HDD failure rate and promptly removed the noisy HDD cooling fans in my Linux box.
Their latest blog post at http://blog.backblaze.com/2013/11/12/how-long-do-disk-drives-last/ has me thinking that some of my colleagues elsewhere that are doing Backblaze like setups should switch to consumer grade HDDs to save on cost.
I do have a 80 Gb Seagate HDD that has survived the years. Admittedly I am not sure what to do with it anymore as it is too small(80 Gb) to be useful and too big (3.5") to be portable. It was used as a main HDD until it's size rendered it obselete hence it's sitting in a USB HDD dock that I use occasionally.
maybe you can find out the age by looking up the serial number but I use the SMART data info that you can see from the Disk Utility in Ubuntu.
 |
| My ancient 3.5" HDD |
 |
| Powered on only for 314 days! |
Hmm pretty low mileage for a 80 Gb HDD eh?
Check out this 320 Gb IDE HDD

Completely anecdotal but I have 3 Seagate 1 Tb HDD dying within a year from a software 5x HDD RAID array from within CentOS. When I checked on the powered on days it says it has been running for 3 years. So I am
1) confused how SMART data records HDD age
2) in agreement with Backblaze that HDD have specific failure phases. (where usage patterns play less of a role perhaps)
3)guessing that most of the data on Backblaze are archival in nature i.e. write once and forget until disaster strikes. So it would be great if Backblaze can 'normalize' the lifespan of the HDD with data access patterns per HDD to make it more relevant for a crowd that has a slightly different usage pattern than pure data archival needs.
That said I think it's an excellent piece of reading if you are concerned about using consumer grade HDD. Kudos the Backblaze team who managed to 'shuck' 5.5 Petabytes of raw HDD to weather the Thailand crisis (wonder how that affected their economics of using consumer grade HDD)
As usual YMMV applies here. Feel free to use consumer grade HDD for your archival needs but be sure to build in redundancy and resilience into your system like the folks in Backblaze.
Monday, 28 October 2013
Illumina Webinar Series: Sequencing Difficult Templates - Why Quality is Everything
Sequencing Difficult Templates - Why Quality is Everything
Date:
Wednesday, November 6
Register Now
Time: 1:00 pm A.E.D.T
Speaker:
Josquin Tibbits, PhD,
Senior Research Scientist, Dept of Environment and Primary Industries
Abstract
For most applications, sequence quality (low error rates, correct library size, even coverage etc), stands out as the key metric for the downstream utility of data from NGS platforms. I investigate the quality and utility of data generated from a range of platforms (454, HiSeq, MiSeq and PGM) for the reference initiated assembly of homopolymer, repeat and low complexity plant plastid genomes. These types of sequences are a good proxy for the more difficult sequence regions found when exploring larger genomes in both agricultural and human sequencing projects. The analysis will show in detail how the different platforms cope with these challenging regions
Wednesday, 2 October 2013
Biome | Q&A with Rich Roberts on single-molecule sequencing technology
The exciting part about single molecule sequencing for me was the ability to sequence low abundance transcripts or have phased haplotypes for human sequencing. Having a high error rate nullifies any advantage in these areas. But I guess it's a tool in the end and how you use it to get meaning results.
"As the previously rapid climb in cost efficiency brought about by next-generation sequencing plateaus, the failure of single-molecule sequencing to deliver might leave some genomics aficionados despondent about the prospects for their field. But a recentCorrespondence article in Genome Biology saw Nobel laureate Richard Roberts, together with Cold Spring Harbor’s Mike Schatz and Mauricio Carneiro of the Broad Institute, argue that the latest iteration of Pacific Biosciences’ SMRT platform is a powerful tool, whose value should be reassessed by a skeptical community.
http://www.biomedcentral.com/biome/rich-roberts-discusses-single-molecule-sequencing-technology/

 Go to article >>
Go to article >>
"As the previously rapid climb in cost efficiency brought about by next-generation sequencing plateaus, the failure of single-molecule sequencing to deliver might leave some genomics aficionados despondent about the prospects for their field. But a recentCorrespondence article in Genome Biology saw Nobel laureate Richard Roberts, together with Cold Spring Harbor’s Mike Schatz and Mauricio Carneiro of the Broad Institute, argue that the latest iteration of Pacific Biosciences’ SMRT platform is a powerful tool, whose value should be reassessed by a skeptical community.
In this Q&A, Roberts tells us why he thinks there’s a need for re-evaluation, and what sparked his interest in genomics in the first place."
http://www.biomedcentral.com/biome/rich-roberts-discusses-single-molecule-sequencing-technology/
Correspondence
The advantages of SMRT sequencing
Genome Biology 2013, 14:405
Go to article >>
Wednesday, 28 August 2013
Case-Based Introduction to Biostatistics with Scott Zeger
For those that might be keen this course is on Coursera
https://www.coursera.org/course/casebasedbiostat
https://www.coursera.org/course/casebasedbiostat
About the Course
The course objective is to enable each student to enhance his or her quantitative scientific reasoning about problems related to human health. Biostatistics is about quantitative approaches - ideas and skills - to address bioscience and health problems. To achieve mastery of biostatistics skills, a student must “see one, do one, teach one.” Therefore, the course is organized to promote regular practice of new ideas and methods.
The course is organized into 3 self-contained modules. Each module except the first is built around an important health problem. The first module reviews the scientific method and the role of experimentation and observation to generate data, or evidence, relevant to selecting among competing hypotheses about the natural world. Bayes theorem is used to quantify the concept of evidence. Then, we will discuss what is meant by the notion of “cause.”
In the second module, we use a national survey dataset to estimate the costs of smoking and smoking-caused disease in American society. The concepts of point and interval estimation are introduced. Students will master the use of confidence intervals to draw inferences about population means and differences of means. They will use stratification and weighted averages to compare subgroups that are otherwise similar in an attempt to estimate the effects of smoking and smoking-caused diseases on medical expenditures.
In the final module, we will study what factors influence child-survival in Nepal using data from the Nepal Nutritional Intervention Study Sarlahi or NNIPPS. Students will estimate and obtain confidence intervals for infant survival rates, relative rates and odds ratios within strata defined by gestational period, singleton vs twin births, and parental characteristics.
The course is organized into 3 self-contained modules. Each module except the first is built around an important health problem. The first module reviews the scientific method and the role of experimentation and observation to generate data, or evidence, relevant to selecting among competing hypotheses about the natural world. Bayes theorem is used to quantify the concept of evidence. Then, we will discuss what is meant by the notion of “cause.”
In the second module, we use a national survey dataset to estimate the costs of smoking and smoking-caused disease in American society. The concepts of point and interval estimation are introduced. Students will master the use of confidence intervals to draw inferences about population means and differences of means. They will use stratification and weighted averages to compare subgroups that are otherwise similar in an attempt to estimate the effects of smoking and smoking-caused diseases on medical expenditures.
In the final module, we will study what factors influence child-survival in Nepal using data from the Nepal Nutritional Intervention Study Sarlahi or NNIPPS. Students will estimate and obtain confidence intervals for infant survival rates, relative rates and odds ratios within strata defined by gestational period, singleton vs twin births, and parental characteristics.
Recommended Background
Interest in the scientific method as broadly related to human health. Ability to reason precisely. Mathematics through pre-calculus.
Suggested Readings
The Mismeasure of Man by Stephen J. Gould is an outstanding resource, but it is not a required text for this course.
Wednesday, 24 July 2013
Illumina produces 3k of 8500 bp reads on HiSeq using Moleculo Technology
Keith blogged about how super long read sequencing methods would be a threat to Illumina in Jan 2013. Today, Illumina can now openly acknowledge the shortcomings of their short reads for various applications like
the reason?
This latest set of data released on BaseSpace
image source: http://blog.basespace.illumina.com/2013/07/22/first-data-set-from-fasttrack-long-reads-early-access-service/
with the integration of Moleculo they have managed to generate ~30 gb of raw sequence data. They have refrained from talking about 'key analysis metrics' that's available in the pdf report. Perhaps it's much easier to let the blogosphere and data scientists dissect the new data themselves.
Am wondering when the 454 versus Illumina Long Reads side-by-side comparison will pop up
so please update me if you see it otherwise I just have to run something on it

These are the files that I have now
total 512M
259M Jul 18 01:01 mol-32-2832.fastq.gz
44K Jul 24 2013 FastTrackLongReads_dmelanogaster_281c.pdf
149K Jul 24 2013 mol-32-281c-scaffolds.txt
44K Jul 24 2013 FastTrackLongReads_dmelanogaster_2832.pdf
151K Jul 24 2013 mol-32-2832-scaffolds.txt
253M Jul 24 2013 mol-32-281c.fastq.gz
md5sums
6845fc3a4da9f93efc3a52f288e2d7a0 FastTrackLongReads_dmelanogaster_281c.pdf
02f5de4f7e15bbcd96ada6e78f659fdb FastTrackLongReads_dmelanogaster_2832.pdf
586599bb7fca3c20ba82a82921e8ba3f mol-32-281c-scaffolds.txt
b25010e9e5e13dc7befc43b5dff8c3d6 mol-32-281c.fastq.gz
6822cfbd3eb2a535a38a5022c1d3c336 mol-32-2832-scaffolds.txt
873f09080cdf59ed37b3676cddcbe26f mol-32-2832.fastq.gz
I have ran FastQC (FastQC v0.10.1) on both samples the images below are from 281c.
you can download the full HTML report here
https://www.dropbox.com/sh/5unu3zba9u21ywj/JT4HdkzfOP/mol-32-281c_fastqc.zip
https://www.dropbox.com/s/mpxa5wx51iqmiz3/mol-32-2832_fastqc.zip
Reading about the Moleculo sample prep method, it seems like it's just a rather ingenious way to stitch short reads which are barcoded to form a single long contig. if that is the case, then I am not sure if the base quality scores here are meaningful anymore since it's a mini-assembly. Also this takes out any quantitative value of the number of reads I presume. So accurate quantification of long RNA molecules or splice variants isn't possible. Nevertheless it's an interesting development on the Illumina platform. Looking forward to seeing more news about it.









Moleculo technology: synthetic long reads for genome phasing, de novo sequencing
CoreGenomics: Genome partitioning: my moleculo-esque idea
Moleculo and Haplotype Phasing - The Next Generation TechnologistNext Generation Technologist
Abstract: Production Of Long (1.5kb – 15.0kb), Accurate, DNA Sequencing Reads Using An Illumina HiSeq2000 To Support De Novo Assembly Of The Blue Catfish Genome (Plant and Animal Genome XXI Conference)
http://www.moleculo.com/ (no info on this page though)
Illumina Announces Phasing Analysis Service for Human Whole-Genome Sequencing - MarketWatch
https://docs.google.com/viewer?url=patentimages.storage.googleapis.com/pdfs/US20130079231.pdf
- assembly of complex genomes (polyploid, containing excessive long repeat regions, etc.),
- accurate transcript assembly,
- metagenomics of complex communities,
- and phasing of long haplotype blocks.
the reason?
This latest set of data released on BaseSpace
 |
| Read length distribution of synthetic long reads for a D. melanogaster library |
The data set, available as a single project in BaseSpace, can be accessed here.
image source: http://blog.basespace.illumina.com/2013/07/22/first-data-set-from-fasttrack-long-reads-early-access-service/
with the integration of Moleculo they have managed to generate ~30 gb of raw sequence data. They have refrained from talking about 'key analysis metrics' that's available in the pdf report. Perhaps it's much easier to let the blogosphere and data scientists dissect the new data themselves.
Am wondering when the 454 versus Illumina Long Reads side-by-side comparison will pop up
UPDATE:
Can't find the 'key analysis metrics' in the pdf report files. Perhaps it's still being uploaded? *shrugs*so please update me if you see it otherwise I just have to run something on it

These are the files that I have now
total 512M
259M Jul 18 01:01 mol-32-2832.fastq.gz
44K Jul 24 2013 FastTrackLongReads_dmelanogaster_281c.pdf
149K Jul 24 2013 mol-32-281c-scaffolds.txt
44K Jul 24 2013 FastTrackLongReads_dmelanogaster_2832.pdf
151K Jul 24 2013 mol-32-2832-scaffolds.txt
253M Jul 24 2013 mol-32-281c.fastq.gz
md5sums
6845fc3a4da9f93efc3a52f288e2d7a0 FastTrackLongReads_dmelanogaster_281c.pdf
02f5de4f7e15bbcd96ada6e78f659fdb FastTrackLongReads_dmelanogaster_2832.pdf
586599bb7fca3c20ba82a82921e8ba3f mol-32-281c-scaffolds.txt
b25010e9e5e13dc7befc43b5dff8c3d6 mol-32-281c.fastq.gz
6822cfbd3eb2a535a38a5022c1d3c336 mol-32-2832-scaffolds.txt
873f09080cdf59ed37b3676cddcbe26f mol-32-2832.fastq.gz
I have ran FastQC (FastQC v0.10.1) on both samples the images below are from 281c.
you can download the full HTML report here
https://www.dropbox.com/sh/5unu3zba9u21ywj/JT4HdkzfOP/mol-32-281c_fastqc.zip
https://www.dropbox.com/s/mpxa5wx51iqmiz3/mol-32-2832_fastqc.zip
Reading about the Moleculo sample prep method, it seems like it's just a rather ingenious way to stitch short reads which are barcoded to form a single long contig. if that is the case, then I am not sure if the base quality scores here are meaningful anymore since it's a mini-assembly. Also this takes out any quantitative value of the number of reads I presume. So accurate quantification of long RNA molecules or splice variants isn't possible. Nevertheless it's an interesting development on the Illumina platform. Looking forward to seeing more news about it.









Other links
Illumina Long-Read Sequencing ServiceMoleculo technology: synthetic long reads for genome phasing, de novo sequencing
CoreGenomics: Genome partitioning: my moleculo-esque idea
Moleculo and Haplotype Phasing - The Next Generation TechnologistNext Generation Technologist
Abstract: Production Of Long (1.5kb – 15.0kb), Accurate, DNA Sequencing Reads Using An Illumina HiSeq2000 To Support De Novo Assembly Of The Blue Catfish Genome (Plant and Animal Genome XXI Conference)
http://www.moleculo.com/ (no info on this page though)
Illumina Announces Phasing Analysis Service for Human Whole-Genome Sequencing - MarketWatch
Illumina Announces Moleculo Long Read Technology and Phasing As Service
First publication using the Long Read Seq (LRseq) The genome sequence of the colonial chordate, Botryllus schlosseri | eLife Contains a diagram explaining the LRSeq protocol. This experiment yielded ~1000 6.3kb fragments
Patent information on the Long Read technologyFirst publication using the Long Read Seq (LRseq) The genome sequence of the colonial chordate, Botryllus schlosseri | eLife Contains a diagram explaining the LRSeq protocol. This experiment yielded ~1000 6.3kb fragments
https://docs.google.com/viewer?url=patentimages.storage.googleapis.com/pdfs/US20130079231.pdf
Friday, 5 July 2013
Windows 8.1 Preview ISOs are available for download
Ah! I didn't know there's Windows 8.1
The ISOs should be helpful if you wish to 'futureproof' your spanking new application in the latest windows or test exisiting apps to see if they might break in the new win8.1
and *cough*usingtheisosasVMsinyourpreferredLinuxenvbutyoukindaneedawindozemachinetodothosetasksthatyoucan'tdoinlinuxcosotherprogrammershaven'theardofbuildingformultiplatformmachines*cough*
well another good reason to use it is that I am pretty sure this ain't happening in Mac or Linux
Microsoft is adding native support for 3D printing as part of the Windows 8.1 update, making it possible to print directly from an app to a 3D printer. The company is announcing the new feature this morning, working with partners including MakerBot Industries, 3D Systems, Afinia, AutoDesk, Netfabb and others.
http://www.geekwire.com/2013/dimension-windows-microsoft-adds-3d-printing-support/
:)

Go http://msdn.microsoft.com/en-us/windows/apps/bg182409 now!
loving the 1.5 Mb/s download here
The ISOs should be helpful if you wish to 'futureproof' your spanking new application in the latest windows or test exisiting apps to see if they might break in the new win8.1
and *cough*usingtheisosasVMsinyourpreferredLinuxenvbutyoukindaneedawindozemachinetodothosetasksthatyoucan'tdoinlinuxcosotherprogrammershaven'theardofbuildingformultiplatformmachines*cough*
well another good reason to use it is that I am pretty sure this ain't happening in Mac or Linux
Microsoft is adding native support for 3D printing as part of the Windows 8.1 update, making it possible to print directly from an app to a 3D printer. The company is announcing the new feature this morning, working with partners including MakerBot Industries, 3D Systems, Afinia, AutoDesk, Netfabb and others.
http://www.geekwire.com/2013/dimension-windows-microsoft-adds-3d-printing-support/
:)

Go http://msdn.microsoft.com/en-us/windows/apps/bg182409 now!
loving the 1.5 Mb/s download here
Wednesday, 3 July 2013
The $1000 myth | opiniomics
*chuckles* I guess only people outside of sequencing needs to be educated on this ..
Obviously, Illumina don't charge themselves list price for reagents, and nor do LifeTech, so it's possible that they themselves can sequence 30x human genomes and just pay whatever it costs to make the reagents and build the machines; but this is not reality and it's not really how sequencing is done today. These guys want to sell machines and reagents, they don't want to be sequencing facilities, plus they still have to pay the staff, pay the bills, make a profit and return money to investors.
http://biomickwatson.wordpress.com/2013/06/18/the-1000-myth/
Tuesday, 2 July 2013
A cloud platform for virtual screening?
Chanced on a blog post inspired by https://www.quantconnect.com/ . Overington suggests the possibility of adopting the model for virtual screening informatics.
For our field I think a SV detection platform would be most 'lucrative'. I would argue that SVs hold an even more important position in contributing to human diversity and disease than the relatively-easier-to-characterize SNPs. I think that #usegalaxy workflow/pipelines might be able to provide a basic framework to build on for people to try to tackle characterization of SVs across multiple sources of sequence data that's publicly available. The problem is who will host the data and platform
The ChEMBL-og - Open Data For Drug Discovery: A cloud platform for virtual screening?: Anyway, have a look, it looks really nice, and got me thinking that it would be a great model for a virtual screening/drug repositioning informatics biotech, where the platform 'brokers' a tournament of approaches coded against data held on a data/compute cloud
For our field I think a SV detection platform would be most 'lucrative'. I would argue that SVs hold an even more important position in contributing to human diversity and disease than the relatively-easier-to-characterize SNPs. I think that #usegalaxy workflow/pipelines might be able to provide a basic framework to build on for people to try to tackle characterization of SVs across multiple sources of sequence data that's publicly available. The problem is who will host the data and platform
The ChEMBL-og - Open Data For Drug Discovery: A cloud platform for virtual screening?: Anyway, have a look, it looks really nice, and got me thinking that it would be a great model for a virtual screening/drug repositioning informatics biotech, where the platform 'brokers' a tournament of approaches coded against data held on a data/compute cloud
Friday, 28 June 2013
Lessons learned from implementing a national infrastructure in Sweden for storage and analysis of next-generation sequencing data
Each time when I change jobs, I will have to go through the adventure (and sometimes pain) to relearn about the computing resources available to me (personal), lab (small sharing pool), and the entire institute/company/school (Not enought to go around usually).
Depending on the job scope / number of cores / length of the job I would then setup the computing resources to run on either of the 3 resources available to me.
Sometimes, grant money appears magically and I am asked by my boss what do I need to buy (ok TBH this is rare). Hence it's always nice to keep a lookout on what's available on the market and who's using what to do what. So that one day when grant money magically appears, I won't be stumped for an answer.
excerpted from the provisional PDF are three points which I agree fully
Three GiB of RAM per core is not enough
you won't believe the number of things I tried to do to outsmart the 'system' just to squeeze enough ram for my jobs. Like looking for parallel queues which often have a bigger amount of RAM allocation. Doing tests for small jobs to make sure it runs ok before scaling it up and have it fail after two days due to insufficient RAM.
MPI is not widely used in NGS analysis
A lot of the queues in the university shared resource has ample resources for my jobs but were reserved for MPI jobs. Hence I can't touch those at all.
A central file system helps keep redundancy to a minimum
balancing RAM / compute cores to make the job splitting efficient was one thing. The other pain in the aXX was having to move files out of the compute node as soon as the job is done and clear all intermediate files. There were times where the job might have failed but as I deleted the intermediate files in the last step of the pipeline bash script, I wasn't able to be sure it ran to completion. In the end I had to rerun the job and keeping the intermediate files
anyway for more info you can check out the below
http://www.gigasciencejournal.com/content/2/1/9/abstract
Depending on the job scope / number of cores / length of the job I would then setup the computing resources to run on either of the 3 resources available to me.
Sometimes, grant money appears magically and I am asked by my boss what do I need to buy (ok TBH this is rare). Hence it's always nice to keep a lookout on what's available on the market and who's using what to do what. So that one day when grant money magically appears, I won't be stumped for an answer.
excerpted from the provisional PDF are three points which I agree fully
Three GiB of RAM per core is not enough
you won't believe the number of things I tried to do to outsmart the 'system' just to squeeze enough ram for my jobs. Like looking for parallel queues which often have a bigger amount of RAM allocation. Doing tests for small jobs to make sure it runs ok before scaling it up and have it fail after two days due to insufficient RAM.
MPI is not widely used in NGS analysis
A lot of the queues in the university shared resource has ample resources for my jobs but were reserved for MPI jobs. Hence I can't touch those at all.
A central file system helps keep redundancy to a minimum
balancing RAM / compute cores to make the job splitting efficient was one thing. The other pain in the aXX was having to move files out of the compute node as soon as the job is done and clear all intermediate files. There were times where the job might have failed but as I deleted the intermediate files in the last step of the pipeline bash script, I wasn't able to be sure it ran to completion. In the end I had to rerun the job and keeping the intermediate files
anyway for more info you can check out the below
http://www.gigasciencejournal.com/content/2/1/9/abstract
Lessons learned from implementing a national infrastructure in Sweden for storage and analysis of next-generation sequencing data
GigaScience 2013, 2:9 doi:10.1186/2047-217X-2-9
Published: 25 June 2013Abstract (provisional)
Thursday, 27 June 2013
Data Analyst - Asia Product Vigilance Procter & Gamble - SG-Singapore-Singapore (Singapore)
Haven't been looking at jobs for a while .. here's something that caught my eye .. typically I feel that pharma jobs are more detailed and pertinent in their hiring criteria since they have a very good idea of what kind of skills they want for the person they are hiring .. it's no different here .. so not much of a surprise. The biggest surprise was that they would only consider a bachelor's degree holder only if they have 8 years of relevant experience. Whether that's equivalent, I shall leave you to decide ..
http://www.linkedin.com/jobs?viewJob=&jobId=5784577
http://www.linkedin.com/jobs?viewJob=&jobId=5784577
Tuesday, 4 June 2013
Bloomberg: MRI for $7,332 Shows Wide Variety in U.S. Medical Costs
There was a kaggle contest for making predictions on patient data to predict return visits if i recall correctly. Wonder what was the aim of this "datapalooza" but this side finding does raises questions about healthcare costing.
Alternatively it might just simply mean that some numbers were wrongly entered into the database. I for one have always been lost in the maze of medical receipts where some items are grouped together and some are separate.
Lesson to be learnt. Important to learn about your primary data.
From Bloomberg, 4 Jun, 2013 4:45:54 AM
The costs of outpatient hospital care vary widely for typical services such as an MRI, according to data released by the U.S. government.
To read the entire article, go to http://bloom.bg/15w43FN
Sent from the Bloomberg iPad application. Download the free application at http://itunes.apple.com/us/app/bloomberg-for-ipad/id364304764?mt=8
Sent from my iPad
Bloomberg: Michael Douglas Oral Sex Cancer Claim Spurs Vaccine Calls
Wow another celebrity "medical advice endorsement"
but it makes sense to immunise guys for the purpose of stemming the spread of the disease and lowering costs of the vaccine for a prevalent disease imho
From Bloomberg, 3 Jun, 2013 8:51:50 PM
Michael Douglas's claim that oral sex led to his throat cancer is spurring calls to vaccinate more boys as well as girls against the human papilloma virus that causes the malignancy.
To read the entire article, go to http://www.bloomberg.com/news/2013-06-03/michael-douglas-oral-sex-cancer-claim-spurs-vaccine-calls.html
Sent from the Bloomberg iPad application. Download the free application at http://itunes.apple.com/us/app/bloomberg-for-ipad/id364304764?mt=8
Sent from my iPad
Friday, 31 May 2013
: [galaxy-user] Newbie: summary or metrics of Galaxy use & adoption?
This will be an interesting thread to watch
---------- Forwarded message ----------
From: "Ramon Felciano" <Feliciano ingenuity.com>
Date: May 31, 2013 2:16 AM
Subject: [galaxy-user] Newbie: summary or metrics of Galaxy use & adoption?
>
Cc:
> Hello all –
>
>
>
> I would like to evaluate Galaxy for integration with Ingenuity's NGS analytics platform, and am trying to pull together some macro materials on how Galaxy has been used and adopted by the community at large. Are there any presentations or wiki pages that summarize basic stuff like:
>
>
>
> # users
>
> # of deployments/ instances (public or private)
>
> Relative use of CloudMan vs locally deployed
>
> Use in any clinical or regulated environments (e.g. CLIA)
>
>
>
> I've seen the materials at http://wiki.galaxyproject.org/Galaxy%20Project/Statistics but this looks to be only related to the public instance. Are there any similar materials that summarize across all installations, perhaps based on user surveys or the like?
>
>
>
> Thanks in advance for your help.
>
>
>
> Ramon
>
>
>
> ____________________________________
>
> Ramon M. Felciano, PhD
>
> Founder and SVP, Research
>
>
>
> INGENUITY Systems, Inc.
>
> 1700 Seaport Blvd., 3rd Floor
>
> Redwood City, CA 94063
>
> 650.381.5100 phone
>
> 650.963.3399 fax
>
>
>
>
>
>
>
>
> ___________________________________________________________
> The Galaxy User list should be used for the discussion of
> Galaxy analysis and other features on the public server
> at usegalaxy.org. Please keep all replies on the list by
> using "reply all" in your mail client. For discussion of
> local Galaxy instances and the Galaxy source code, please
> use the Galaxy Development list:
>
> http://lists.bx.psu.edu/listinfo/galaxy-dev
>
> To manage your subscriptions to this and other Galaxy lists,
> please use the interface at:
>
> http://lists.bx.psu.edu/
>
> To search Galaxy mailing lists use the unified search at:
>
> http://galaxyproject.org/search/mailinglists/
Thursday, 30 May 2013
bash script to timestamp snapshot backups of directory
modified a script from
http://aaronparecki.com/articles/2010/07/09/1/how-to-back-up-dropbox-automatically-daily-weekly-monthly-snapshotsto be a simplified snapshot copy of the FOLDER and append the date to the folder name. Useful to have an backup on Dropbox since some files might go missing suddenly on shared folders.
#!/bin/bash
#eg.
#sh snapshot.sh FOLDER [go]
# 'go' will execute
path=Snaphotbackup-`date +%m-%b-%y`
# Run with "go" as the second CLI parameter to actually run the rsync command, otherwise prints the command that would have been run (useful for testing)
if [[ "$2" == "go" ]]
then
rsync -avP --delete $1 $1-$path
else
echo rsync -avP --delete $1 $1-$path
fi
Tuesday, 21 May 2013
Evaluation of autosomal dominant retina - PubMed Mobile
http://www.ncbi.nlm.nih.gov/m/pubmed/23687434/
METHODS: Publicly available data from the Exome Variant Project were analyzed, focusing on 36 genes known to harbor mutations causing autosomal dominant macular dystrophy.
RESULTS: Rates of rare (minor allele frequency ≤0.1%) and private missense variants within autosomal dominant retinal dystrophy genes were found to occur at a high frequency in unaffected individuals, while nonsense variants were not.
CONCLUSIONS: We conclude that rare missense variations in most of these genes identified in individuals with retinal dystrophy cannot be confidently classified as disease-causing in the absence of additional information such as linkage or functional validation.
PMID 23687434 [PubMed - as supplied by publisher]
Saturday, 18 May 2013
How does your bash prompt look like?
decided to add color and full path to my bash prompt to make it easier for me to see which ssh window belongs to where absolute life saver at times when you are running several things and the tabs just don't show enough info
Plus having the user@host:/fullpath/folder in your bash prompt makes it easy to copy and paste a 'url' to scp files into that folder directly instead of typing from scratch The only downside to this is if you are buried deep in folders, you are not going to have a lot of screen estate left to type long bash commands without confusing yourself
My final choice:
PS1="\[\033[35m\]\t\[\033[m\]-\[\033[36m\]\u\[\033[m\]@\[\033[32m\]\h:\[\033[33;1m\]\w\[\033[m\]\$ "

Source:
8 Useful and Interesting Bash Prompts - Make Tech Easier
http://www.maketecheasier.com/8-useful-and-interesting-bash-prompts/2009/09/04
How to: Change / Setup bash custom prompt (PS1)
http://www.cyberciti.biz/tips/howto-linux-unix-bash-shell-setup-prompt.html
Thursday, 16 May 2013
Framework for evaluating variant detection methods: comparison of aligners and callers | Blue Collar Bioinformatics
Useful info!
Brad Chapman wrote a lengthy blog post doing a recent comparison of 3 variant callers
- FreeBayes (0.9.9 296a0fa): A haplotype-based Bayesian caller from the Marth Lab, with filtering on quality score and read depth.
- GATK UnifiedGenotyper (2.4-9): GATK's widely used Bayesian caller, using filtering recommendations for exome experiments from GATK's best practices.
- GATK HaplotypeCaller (2.4-9): GATK's more recently developed haplotype caller which provides local assembly around variant regions, using filtering recommendations for exomes from GATK's best practices.
http://bcbio.wordpress.com/2013/05/06/framework-for-evaluating-variant-detection-methods-comparison-of-aligners-and-callers/
This evaluation work is part of a larger community effort to better characterize variant calling methods. A key component of these evaluations is a well characterized set of reference variations for the NA12878 human HapMap genome, provided by NIST's Genome in a Bottle consortium. The diagnostic component of this work supplements emerging tools like GCAT (Genome Comparison and Analytic Testing), which provides a community platform for comparing and discussing calling approaches.
I'll show a 12 way comparison between 2 different aligners (novoalign and bwa mem), 2 different post-alignment preparation methods (GATK best practices and the Marth lab's gkno pipeline), and 3 different variant callers (GATK UnifiedGenotyper, GATK HaplotypeCaller, and FreeBayes). This allows comparison of openly available methods (bwa mem, gkno preparation, and FreeBayes) with those that require licensing (novoalign, GATK's variant callers). I'll also describe bcbio-nextgen, the fully automated open-source pipeline used for variant calling and evaluation, which allows others to easily bring this methodology into their own work and extend this analysis.
Wednesday, 15 May 2013
A Next-Generation Sequencing Method for Genotyping-by-Sequencing of Highly Heterozygous Autotetraploid Potato.
PLoS One. 2013 May 8;8(5):e62355. doi: 10.1371/journal.pone.0062355. Print 2013.
A Next-Generation Sequencing Method for Genotyping-by-Sequencing of Highly Heterozygous Autotetraploid Potato.
Source
Laboratory of Plant Breeding, Wageningen University, Wageningen, The Netherlands ; The Graduate School for Experimental Plant Sciences, Wageningen, The Netherlands.
Abstract
Assessment of genomic DNA sequence variation and genotype calling in autotetraploids implies the ability to distinguish among five possible alternative allele copy number states. This study demonstrates the accuracy of genotyping-by-sequencing (GBS) of a large collection of autotetraploid potato cultivars using next-generation sequencing. It is still costly to reach sufficient read depths on a genome wide scale, across the cultivated gene pool. Therefore, we enriched cultivar-specific DNA sequencing libraries using an in-solution hybridisation method (SureSelect). This complexity reduction allowed to confine our study to 807 target genes distributed across the genomes of 83 tetraploid cultivars and one reference (DM 1-3 511). Indexed sequencing libraries were paired-end sequenced in 7 pools of 12 samples using Illumina HiSeq2000. After filtering and processing the raw sequence data, 12.4 Gigabases of high-quality sequence data was obtained, which mapped to 2.1 Mb of the potato reference genome, with a median average read depth of 63× per cultivar. We detected 129,156 sequence variants and genotyped the allele copy number of each variant for every cultivar. In this cultivar panel a variant density of 1 SNP/24 bp in exons and 1 SNP/15 bp in introns was obtained. The average minor allele frequency (MAF) of a variant was 0.14. Potato germplasm displayed a large number of relatively rare variants and/or haplotypes, with 61% of the variants having a MAF below 0.05. A very high average nucleotide diversity (π = 0.0107) was observed. Nucleotide diversity varied among potato chromosomes. Several genes under selection were identified. Genotyping-by-sequencing results, with allele copy number estimates, were validated with a KASP genotyping assay. This validation showed that read depths of ∼60-80× can be used as a lower boundary for reliable assessment of allele copy number of sequence variants in autotetraploids. Genotypic data were associated with traits, and alleles strongly influencing maturity and flesh colour were identified.
- PMID:
- 23667470
- [PubMed - in process]
Jolie Has Double Mastectomy to Thwart Cancer Gene Risk - Bloomberg
This will have a ripple effect on genetic testing ... mastectomy ... I have no clue .. I wonder if it's possible to find the exact BRCA1 gene variant that she carries. Quite curious to see the literature on a gene variant that will make non-science people decide on invasive surgery for a '90% protection' against cancer.
I wonder if it's really too late to do the mastectomy after you discover you have cancer ..
Original article
http://www.bloomberg.com/news/2013-05-14/jolie-has-double-mastectomy-to-thwart-cancer-gene-risk.html
http://www.bloomberg.com/news/2013-05-14/jolie-has-double-mastectomy-to-thwart-cancer-gene-risk.html
What's the gene that led to Angelina Jolie's double mastectomy? - CNN.com
Opinion: Angelina Jolie's brave message - CNN.com
http://edition.cnn.com/2013/05/14/opinion/caplan-angelina-jolie-mastectomy/index.html
Opinion: Jolie's choice carries risks with benefits - CNN.com
Friday, 10 May 2013
PLOS ONE: A Multi-Platform Draft de novo Genome Assembly and Comparative Analysis for the Scarlet Macaw (Ara macao)
http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0062415
A multi platform approach to whole genome sequencing of novel genomes is the best way forward. This is quite a comprehensive paper stating the steps done to achieve the final assembly with annotations.
Interestingly CLC bio was the software used for the assembly.
It would be interesting if the assembly was 'multi platform' as well.
Sunday, 5 May 2013
Article: Stranger Visions: DNA Collected from Found Objects Synthesized to Create 3D Printed Portraits
This is kinda creepy even to someone like me.
Having your DNA sequenced (or rather typed) by a stranger to create approximate portraits of you.
While no sinister intent is present here. It should scare the hell outta the man on the street. (Hopefully the gum spitter and butt thrower variant )
No doubt the project does cast human genomics in a bad light ( privacy issues) but I think a watchful public eye is good for publicly funded science.
Stranger Visions: DNA Collected from Found Objects Synthesized to Create 3D Printed Portraits
http://www.thisiscolossal.com/2013/05/stranger-visions-dna-collected-from-found-objects-synthesized-to-create-3d-printed-portraits/
Sent via Flipboard
http://www.thisiscolossal.com/2013/05/stranger-visions-dna-collected-from-found-objects-synthesized-to-create-3d-printed-portraits/
Sent via Flipboard
Sent from myPhone
Monday, 29 April 2013
Watch "Illumina on AWS - Customer Success Story" on YouTube
Illumina was featured in AWS customer success story recently.
I was initially surprised at the fact that Miseq data is the majority of the data analyzed by users of Basespace (it was mentioned in the video that Illumina is beginning to hook Hiseq machines to Basespace)
I was less surprised at their claims that 90% of DNA bases sequenced were performed on Illumina machines
https://www.youtube.com/watch?v=sf9LGG-CZUw&feature=youtube_gdata_player
Illumina, a California-based leading provider of DNA sequencing instruments, uses AWS to enable researchers to process and store massive amounts of data in the AWS Cloud. AWS offers the scalability and power in a secure environment that Illumina needs to help researchers collaborate while sequencing and analyzing large amounts of data.
Watch the Video »
Saturday, 27 April 2013
Amazon.com: Bayesian Computation with R (Use R) eBook: Jim Albert: Kindle Store
The first edition Kindle ebook is free (source R bloggers) but unfortunately not for Asia Pacific region
The R scripts from the book (2nd edition but there's a lot of overlap) can be obtained on Jim Albert's web site http://bayes.bgsu.edu/bcwr/
Tuesday, 23 April 2013
Life Technologies hiring software engineers with cloud computing experience
http://lifetech.tmpseoqa.com/articles/tech
Life Technologies is also moving into cloud computing. Would be interesting to see if they can bring anything new to the rather crowded table.
Life Technologies is also moving into cloud computing. Would be interesting to see if they can bring anything new to the rather crowded table.
Thursday, 11 April 2013
Cufflinks 2.1.0 released
From: Cole Trapnell <cole at cs.umd.edu>
Date: 11 April, 2013 12:52:18 AM GMT+08:00
To: "bowtie-bio-announce
Subject: [Bowtie-bio-announce] Cufflinks 2.1.0 released
2.1.0 release - 4/10/2013
This release substantially improves the accuracy, speed, and memory footprint of Cufflinks and Cuffdiff. It is recommended for all users. Those who wish to see the impact of the accuracy improvements should look at the new benchmarking section. In addition to numerous bugfixes, the main changes are as follows:
- Cuffdiff now includes a new statistical test. Prior versions used a delta method-based test, which lacked power for designs with more than a few replicates. The new test directly samples from the beta negative binomial model for each transcript in each condition in order to estimate the null distribution of its log fold change under the null hypothesis. This test is substantially more powerful, resulting in improved accuracy over all experimental designs, particularly those with more than three replicates. A similarly improved test is now used for isoform-switching. The benchmarking page shows the improvements in detail.
- Prior versions of Cuffdiff reported the FPKM for each gene and transcript that maximizes the joint likelihood of the reads from all replicates pooled together. In version 2.1, Cuffdiff instead reports the mean of the maximum likelihood estimates from each replicate processed independently. As shown in the benchmarking section, these two methods report nearly identical values. However, the new method is faster and simpler to compute, and will enable new features for future releases.
- The high and low confidence intervals reported by Cufflinks and Cuffdiff are now constructed from the samples generated from the beta negative binomial model, rather than estimated as twice the standard deviation. This better reflects the underlying distribution of the FPKM.
- The library normalization system in Cuffdiff 2 has been overhauled, and several new normalization-related options have been added:
- The new --library-norm-method option now sets which method should be used to compute scaling factors for the library sizes. The default method geometric is the same as prior releases of Cuffdiff (and the same as DESeq). The optional modes quartile and classic-fpkm are also supported.
- The new --dispersion-method option controls how the variance model should be computed for each condition. The default mode pooled computes a mean-variance model for each condition that has multiple replicates, averages these models to generate a "pooled" average, and uses it for all conditions. This policy is borrowed from DESeq. Alternative models blind and per-condition are also supported. Prior versions of Cuffdiff used the method per-condition.
- Several bugs for quartile normalization have been fixed.
- Quartile normalization is no longer supported in Cufflinks, just in Cuffdiff. Cufflinks only supports the classic-fpkm mode.
- All library size normalization is now conducted through the internal scaling factor. The external scaling factor should always be set to 1.0.
- Library sizes and dispersions are now computed only on fragment counts from compatible fragments. Prior versions counted intronic and other structurally incompatible fragments in some sections of the code.
- An optimized sampling procedure drastically improves running time for Cuffdiff. Cufflinks also benefits from this change. The improvements are particularly noticeable on deeply sequenced libraries.
- The range of p-values that users should expect from Cuffdiff has changed. Because the test is now based on explicit sampling from the beta negative binomial, users will not see values less than 10^-5 by default. The test_stat field of Cuffdiff's output still contains the delta method's test statistic, but this test statistic is not used to compute p-values. It is preserved for backward compatibility with some functions in CummeRbund.
- Some extraneous temporary output files have been removed after Cuffmerge runs.
--------------------------------------------------------------
_______________________________________________
Bowtie-bio-announce mailing list
Bowtie-bio-announce@lists.sourceforge
https://lists.sourceforge.net/lists/listinfo/bowtie-bio-announce
Tuesday, 2 April 2013
TIL 'with open' in Python
I have always delayed learning about writing better code and what you don't know you often won't put into practice but when I do come across tips and optimizations, I use them heavily.
I am slightly embarrassed to say that today I learnt about 'with open' for reading files in python from Zen of Python.
Read From a File
Learn to Program: Crafting Quality Code
I am slightly embarrassed to say that today I learnt about 'with open' for reading files in python from Zen of Python.
Read From a File
Use the with open syntax to read from files. This will automatically close files for you.
Bad:
Good:
The with statement is better because it will ensure you always close the file, even if an exception is raised.
There's some good reading on styles and preferred 'Pythonic' ways of coding if you follow the code style guide in The Hitchhiker's Guide to Python.
One other good source is the plethora of online courses available now on Coursera or other sites e.g.
Learn to Program: Crafting Quality Code
Subscribe to:
Posts (Atom)